1. Synthetic organic chemistry
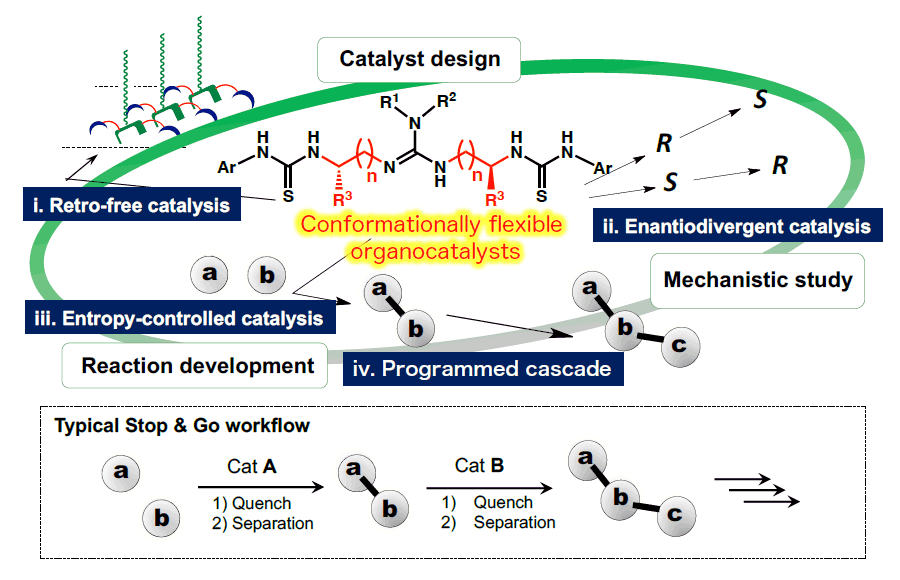
1-1) Dynamic asymmetric organocatalysis
Asymmetric catalysis is one of the most powerful methodologies for engineering chiral molecules, but most applications have relied on the strategy of using structurally defined chiral templates to reduce the number of transition states. In our work, we have developed a new class of asymmetric organocatalysts capable of multiple catalytic functions through the incorporation of a conformationally flexible chiral spacer. Using these new organocatalysts, we can achieve different reactions within the same reaction vessel simply by applying the appropriate external stimuli to trigger the desired catalytic function. This versatility allows us to explore several dynamic catalytic processes, including i) retro-free catalysis, ii) sequential enantio-divergent catalysis, iii) entropy-controlled catalysis, and iv) programmed-cascade. Furthermore, we have revealed an entropic mechanism for controlling selectivity that is similar to that found in enzymes, in which selectivity increases as the temperature rises.
Reviews: Chem. Commun. 2012, 48, 7777–7789; J. Synth. Org. Chem. Jpn. 2015, 798–809.
Representative papers: Angew. Chem. Int. Ed. 2010, 49, 7299–7303; Angew. Chem. Int. Ed. 2010, 49, 9254–9257.
1-2) Transition metal catalysis
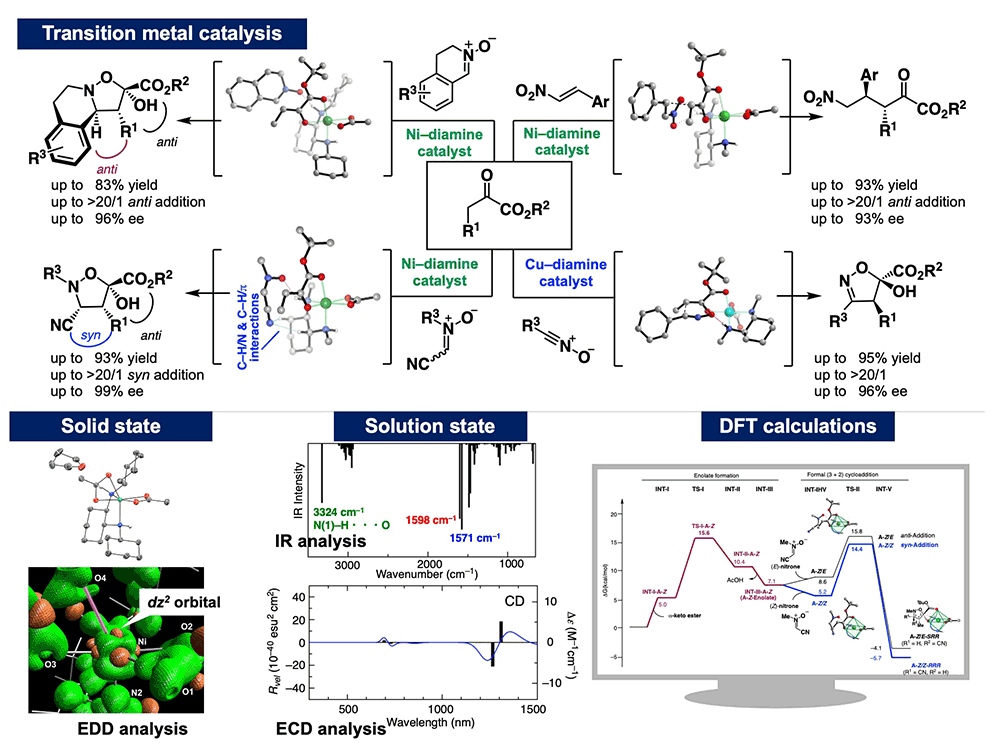
Significant advances in research on transition metal catalysts have been made by exploring their unique higher-order structures resulting from the splitting of d-orbitals. However, finding effective catalysts traditionally has relied on a trial-and-error process of adjusting the metal salts and chiral ligands based on the reaction yield and selectivity, and the mechanisms by which these transition metal complex catalysts activate substrates often remain a black box.
In our research, we have elucidated the electronic structure of the ground state of an original Ni(II)–diamine complex developed in our laboratory. By combining experimental and computational chemistry, we have proposed a new transition state model featuring dual activation: enolate formation on metal centrochirality (inner-sphere activation) and H-bonding activation (outer-sphere activation). This research offers new guidelines for catalyst design, paving the way for the development of unprecedented catalytic transformations. Using these results, we have so far developed several methods for providing architecturally complex molecules with contiguous chiral centers.
Reviews: Synlett 2020, 6, 523–534; Farumashia 2019, 55, 934–938.
Representative papers: Nat. Commun. 2017, 8, 14875; J. Am. Chem. Soc. 2017, 139, 8661–8666; J. Am. Chem. Soc. 2021, 143, 9094–9104.
1-3) Cross-couplings of persistent radicals
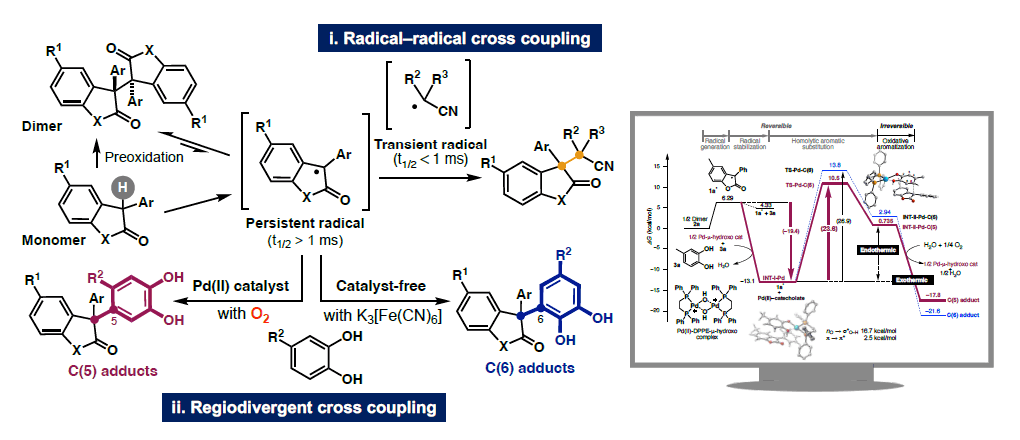
New mechanistic principles are crucial for the design of reactions that will enable efficient synthesis of molecules unlike anything seen to date. To achieve this goal, we are exploring radical chemistry in addition to the aforementioned enolate chemistry. In this study, we have established an atom-economical method for radical generation that produces persistent radical species with lifetimes (t1/2) of milliseconds or longer from 2-oxyindole and benzofuranone dimers. By introducing the “lifetime” of the active species into our reaction design, we have achieved i) radical–radical cross-coupling with reactive radical species derived from azo compounds (t1/2 < milliseconds), and ii) regio-divergent cross-coupling with catechols. Furthermore, we have demonstrated a unique energy profile involving anion/radical coupling between Pd-catecholate (HOMO) and a persistent radical (SOMO) [homolytic aromatic radical substitution reaction (energetically uphill reaction)] followed by oxidative aromatization (downhill reaction), ultimately converting to more stable products.
Reviews: Pure Appl. Chem. 2024, 96, 5-21; Bull. Chem. Soc. Jpn. 2021, 94, 1066–1079.
Representative papers: ACS Catal. 2020, 10, 12770–12782; Angew. Chem. Int. Ed. 2024, e202405876.